


What's Alprazolam's Deal?
An investigation into the Rollercoaster of Benzodiazepines

I’ve been informed by one of my good friends, Larry (Hi Larry!) who reads this blog that my writing needs more “fun personal anecdotes.” He also suggests that “maybe a cat photo or two wouldn’t hurt.”
I’m fresh out of fun personal anecdotes at the moment, but I do have plenty of cat photos, so here are two:


Happy, Larry?
If you’re been doing psychiatry for any amount of time I guarantee that at some point you come across someone who tells you that under no circumstances should you ever consider using alprazolam (Xanax). The primary reason being that alprazolam is very short acting, which results in a rapid diminishment of effect that leaves patients (particularly anxious patients who are susceptible to becoming distressed during any type of change) feeling worse than when they started.
Why? Well, that’s very simple. You see, alprazolam has a very short half-life.
Oh hello rhetorical foil, nice to see you. Here’s the thing, it doesn’t, actually. Alprazolam’s mean half-life (λ) in healthy individuals is 11.2h (range 6.3-26.9h), which isn’t appreciably different from lorazepam (Ativan) λ = 12h, or oxazepam λ = 8h.
You might think that lorazepam has some longer acting metabolite(s) to explain the difference, but its primary metabolic product is the pharmacologically inactive lorazepam glucuronide. Alprazolam on the other hand does have active metabolites — 4-hydroxyalprazolam and α-hydroxyalprazolam — though most alprazolam is eliminated in the urine unchanged. Only 10% is converted into the active metabolites.
Well, it’s not just that, alprazolam also has a uniquely fast rate of absorption!
Let’s compare it to the T-max1 of lorazepam and oxazepam:
Alprazolam = ~1.8h
Lorazepam = ~2h
Oxazepam = ~3h
So, modestly faster than oxazepam, almost identical to lorazepam.
Ok, I have some citations now. Its one of the most lipophilic benzodiazepines, which means that it crosses the blood brain barrier fast and acts quickly! Er… wait a sec, this source says it has relatively low lipophilicity, so that means that it gets eliminated more quickly?
Yeah… why don’t we just look at the primary experimental literature?
D.J. Greenblatt — in addition to dropping sick beats in the OR2 — also dropped this paper in the British Journal of Anesthesiology in 1983 (ft. et al). His group analyzed 14 individual benzodiazepines using retention time in reverse-phase HPLC3 and partition ratios in aqueous buffer + octanol solution as in vitro proxies for lipophilicity.
As a little refresher for those who have (rightly) memory-holed their experiences in organic chemistry:
Partition ratios tell you the relative solubility of a solute in two different solvents. Octanol is an amphiphilic solvent that approximates the characteristics of lipids in vivo, and so the octanol:buffer partition ratio acts as a decent proxy for lipophilicity. The higher the ratio, the higher the lipophilicity.
In reverse-phase HPLC, you take a small amount of your dissolved solute and put it into a tube filled with silica particles coated with a hydrophobic surface. Then you wash the silica particles with your choice of solvent (different solvents can be used for different compounds) and use a device to monitor the amount of the solvent in the solution exiting the tube. Retention time is the time it takes to reach peak concentration at the detector.
Sorry to retraumatize you. Here are the results from Greenblatt’s paper:

In the octanol:buffer experiment, alprazolam is clearly not very lipophilic relative to the other benzodiazepines. In the HPLC condition, it is solidly in the middle of the pack. The question is, which of these models best mimics in vivo observations?
Well, the paper looked at that too in “a series of healthy male volunteers aged less than 40 yr who were within 10% of ideal body weight.” No demographics provided, the 1980s were a simpler time. Anyway, here are the results:

We can see that both methods predict in vivo distribution pretty well, and alprazolam hangs out in the same part of the graph. Regardless of which model you choose, the obvious point is that alprazolam is either much less lipophilic than most of the other common benzodiazepines, or in the same neighborhood as lorazepam.
Hmm, I see that alprazolam’s unbound volume of distribution is pretty low relative to everything else. Could that have something to do with it?
Maybe?
First, remember that volume of distribution (Vd) is calculated as follows:
The larger the number, the more extensively a drug is distributed into peripheral tissues.
Keen-eyed readers will note that we’re talking about the unbound volume of distribution (uVd) (i.e. the amount of drug not bound to proteins in the blood), which is important in this context because only unbound drug is available for distribution outside of the vascular system. Just relying on Vd can be very misleading for drugs like diazepam which is >95% protein bound in circulation. If you just relied on UpToDate, which gives a Vd for diazepam of 0.6-1.8 L/kg, you’d think it should behave an awful lot like alprazolam whose Vd is listed as 0.84-1.42 L/kg. As you can see in the Greenblatt paper, diazepam’s uVd is (eyeballing here) ~90 L/kg, while alprazolam’s is (again, eyeballing) ~5 L/kg.
What does this prove? Well, I think this is a point in favor of the hypothesis that alprazolam is not particularly lipid soluble. It also suggests that — as we’ll see later — even though alprazolam and diazepam behave similarly to one another, the explanations for why are probably different.
Hm, what about some other pharmacodynamic stuff? Is there something weird about how quickly it gets into the CSF?
Unfortunately, things only get more confusing from here. In another Greenblatt paper from 1983, In vitro correlates of benzodiazepine cerebrospinal fluid uptake, pharmacodynamic action and peripheral distribution, the authors use anesthetized male cats look at the kinetics of 8 benzodiazepines in the CSF and the relationship to onset and duration of slow-wave EEG activity, a classic finding of benzodiazepine administration.
There does appear to be a direct relationship between lipophilicity, CSF entry half-life (time to 50% of CSF peak), and time to peak CSF concentrations, but it (seems) quite modest. Alprazolam was the longest in both CSF entry half-life (i.e. time to 50% of maximal CSF concentration, ~4 minutes) and peak CSF concentrations (~30 minutes). Others were significantly shorter: Lorazepam (entry half-life: 1.6min, peak: 7min), Diazepam (entry half-life: 0.44min, peak: 3.7min).
All 8 benzodiazepines had CSF elimination half-lives that were nearly identical to their plasma elimination half-lives, as you can see here:

Clearly this isn’t some strange issue where alprazolam is rapidly removed from CSF more quickly than its peers (e.g. by some transporter protein).
The EEG findings are no more enlightening. The onset of EEG slowing was rapid and all benzos induced changes within 4 minutes. Duration of slowing was inversely correlated with unbound Vd. In other words, the less lipophilic the drug -or- the more free drug available in the plasma/CSF, the longer it tends to induce EEG changes. This might seems a bit counterintuitive, but makes some sense to me. Anesthetics that are highly lipid soluble usually have a fast onset and offset of action because concentrations fall rapidly after administration as the drug is distributed into other tissues.

As you can see above, plasma (and therefore CSF elimination) is not correlated with the duration of slow-wave activity. What appears to matter here is blood concentration.
The problem is, again, alprazolam and lorazepam really don’t look that different in terms of the duration of slow-wave activity. It’s only a 7 minute difference (35 vs. 28 minutes).
Yeah, not getting anywhere here, huh? I give up — you’ll have to continue this narrative without me.
No problem rhetorical foil, until next time.
You know, maybe this whole thing about alprazolam having a fast onset/offset isn’t even real. Maybe it’s just another pharmacological myth, like trazodone being effective for insomnia or antihistamines clearly being deliriogenic. I haven’t actually seen any papers that have documented a differential effect concretely.
Hey, uh, I’m back and I found this paper: Comparative pharmacokinetics and pharmacodynamics of lorazepam, alprazolam and diazepam from Ellinwood et al.. You might want to take a look at the first couple paragraphs:
Benzodiazepine pharmacodynamic profiles are not predicted well by dispositional pharmacokinetics following acute dosing. The offset of diazepam-induced psychomotor impairment is much more rapid than the corresponding decline of concurrent bound and unbound diazepam concentration. In contrast, lorazepam has a prolonged period of impairment relative to diazepam which is clearly not a function of drug elimination rates, since the elimination half-life of lorazepam is one-third that of diazepam.
Either differential absorption-distribution pharmacokinetics (Arendt et al. 1983) or receptor kinetics appear to be a more likely explanation of the observed differences in pharmacodynamics between the two drugs than their elimination kinetics.
Sometimes you read something that makes you feel — as the kids say — seen.
First a little about study design. Ellinwood and co. used 8 human males between ages 21-26 that were within 10% of ideal body weight, and trained them on a series of cognitive-neuromotor tasks that included continuous subcritical tracking (SCT) and digit symbol substitution (DSS); it’s not terribly important to know the exact nature of these tasks, other than that the CST is a motor coordination task, and the DSS is a cognitive task. On test days, participants were dosed (strongly!) with one of the three benzodiazepines or placebo, after which the aforementioned cognitive tests were given at specified times alongside blood draws to measure blood levels.

These graphs show how the performance on the two tests varied alongside serum levels over time, as a percentage of the “observed maximum.” For the tests, “maximum” refers to the maximum difference between their baseline test scores and scores after taking the benzodiazepine. (I don’t really understand why neither graph has points that hit 100%, I think because they are aggregates of all of the test subjects)
What is interesting here is the relationship (or the lackthereof) between serum levels and performance over time. Notice how serum levels and performance move together pretty closely in the first 100-120 minutes or so, but then task performance rapidly improves even though serum levels stay relatively high? This indicates some sort of rapid tolerance effect with both alprazolam and diazepam.
When we look at the lorazepam graph, however, we see a very different story. A much more gradual return to baseline.

This is even better visualized with the graphs overlaid. First for the eye-tracking test:
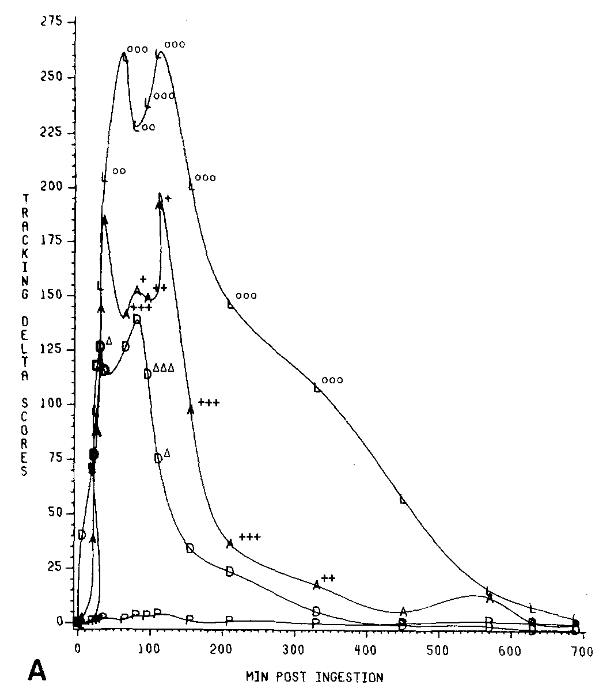
And then for the number substitution test:

Even though it’s lorazepam and alprazolam that have the similar half-lives, it’s actually diazepam and alprazolam that show the same rapid offset of psychomotor effects.
Alprazolam does still behave a bit differently from diazepam though, in that its onset is totally divorced from serum levels. It’s a little hard to see, though, so let’s use some different graphs that show how participants performed on the CST over time.

The horizonal axis shows serum concentrations of the drug, the vertical axis shows the delta between baseline CST score and the score at the timepoint on the graph; higher numbers means worse performance relative to baseline. The numbers that appear next to the line indicate the time the score was collected in minutes post-dosing. Don’t worry about the dashed line, it’s not super important for this discussion.
Notice how there’s basically nothing going on until that 20 minute mark, and then it’s just like a rocket upwards, even though serum concentrations aren’t changing at all?
Now, let’s look at the graph for diazepam:
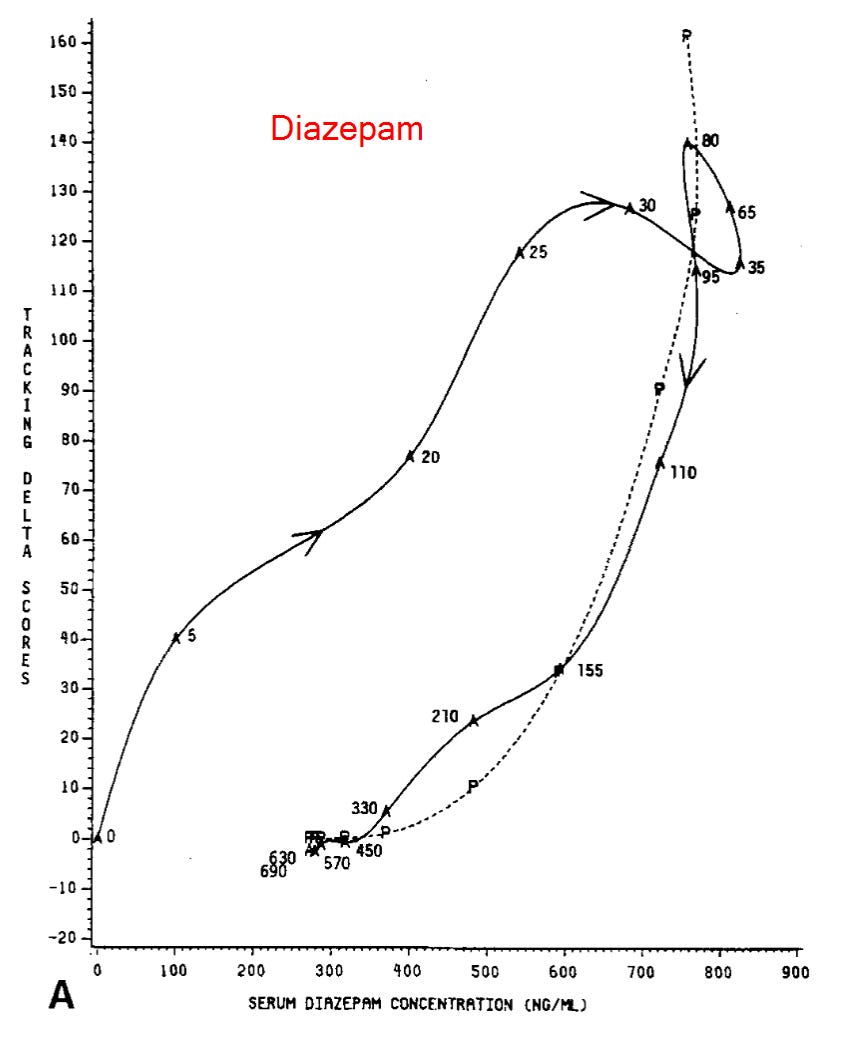
The difference is pretty striking, no? What might explain this? Do you remember what it was about alprazolam that was from the Greenblatt paper above? The (what I now regret calling “modest”) time to peak CSF concentration of about 30 minutes, much shorter than lorazepam (~7min) or diazepam (~3.7min). The authors of this paper propose that this may be exactly what explains the difference.
However, there is still the question of what is going on to create such a rapid diminution of alprazolam’s action. Everything we have found so far indicates that it is obviously not explained by CSF elimination kinetics. The Ellinwood paper points out that lorazepam and alprazolam have pretty similar binding affinities, so it’s probably not that either. Instead, they suggest two hypotheses to explain why diazepam (and maybe alprazolam) behave differently than lorazepam.
First, they propose that receptor association-dissociation rate constants play a role. They cite a couple papers that have found that diazepam’s dissociation rate from GABA receptors is 9-10x faster than lorazepam. This, they suggest, lines up with the findings in this paper, which show that eye tracking impairment leads serum levels for diazepam and lags serum levels for lorazepam. They also point out that, unlike diazepam or alprazolam, lorazepam’s chemical structure has:
a chloride ion ion the ortho position of the ‘C’ ring, which has been associated with the very high affinity of these benzodiazepines and may account for a specific long-lasting lorazepam effect.
Second, they speculate that differential binding weirdness at the GABA-A receptors may be at play here. Considering just how complicated receptor-ligand interactions can be (see: functional selectivity, receptor heterodimerization) and the sheer diversity of GABA-A receptor subtypes (there are at least 26!), I find this entirely plausible.
In fact, this is not just speculation. Take this paper from McKernan et al. (2000). They used genetically modified mice to show that modifications to the α1-subunit of the GABA-A receptor altered diazepam’s sedating effects, but not its anxiolytic effects. For a deeper dive, I recommend this review by Sigel and Ernst.
This is the point at which I actually, seriously, truly, planned to wrap up this essay, but then one Dr. D.J. Greenblatt responded to the email I sent him asking what he thought explained this alprazolam business. Here is the relevant part of his email response on the topic:
There is more to this than half-life. The other issues are: rate of absorption, and rate of entry into the CNS.
Alprazolam is absorbed more rapidly than lorazepam, producing a faster onset of effect and therefore a reinforcement "buzz" after each dose (see Greenblatt 1988). This seems to be associated with abusability.
In the case of the 3-OH benzodiazepines (lorazepam, oxazepam), the -OH substitution reduces lipophilicity, and slows CNS entry. Together with the slow absorption rate, this tends to reduce abusability. The slow CNS entry can be seen both in clinical and experimental studies (Greenblatt 1989 and Greenblatt & Sethy 1990). Not so for alprazolam, which does not have the -OH substitution, and gets into the brain relatively fast (Venkatakrishnan 2005).
I thought these papers provide a good story for the differences between diazepam and lorazepam, but I don’t think that the Venkatakrishnan paper provides evidence that should make us assume that alprazolam mimics diazepam.
I would be willing to make the assumption if alprazolam was obviously very lipophilic like diazepam, but remember that Greenblatt 1983 paper. Alprazolam was second to last in partition ratios (18 vs. diazepam's 309) and in the middle of the pack in the reverse-phase HPLC. Alprazolam’s retention time was just 2.5 minutes longer than lorazepam, and 12.5 minutes shorter than diazepam’s. It also had the lowest unbound volume of distribution of any of the other benzodiazepines tested.
Also remember that the Arendt et al. 1983, shows alprazolam as the slow one when it comes to CNS penetration. Mean time to peak CSF concentration was 28 minutes, while lorazepam (7 min) and diazepam (3.7 min) look blazingly fast in comparison. There is strange discordance here with onset of slow-wave EEG activity, where alprazolam showed onset of slow-wave EEG activity within ~30 seconds, while lorazepam took about 4 minutes.
I sent Dr. Greenblatt back these observations to see what his interpretation was, but I had not yet heard back from him at the time I published this. If he does, I will update here.
We Learned Something, I Guess?
Ok, yes we actually did, or at least I did. The alprazolam fast onset and offset really does seem to be a thing, not just another psychopharmacological legend. That is… actually the most clinically useful thing I’ve come across through this whole process.
I also learned that diazepam has a very similar clinical effect. Not something that I would’ve supposed at all given how much we talk about diazepam as a drug with a very long half-life!
Overall, this bit of research was (yet another) lesson that pharmacology is way, way more complicated than we think. I’m starting to think that trying to use pharmacokinetic/pharmacodynamic properties to predict clinical effect is almost exactly backwards, instead of just a little backwards. Those poor shmucks in the pharmaceutical industry have no other choice, but at least we do.
T-max is time to peak serum level
Check out his sophomore album Sevo-lution, a follow up to his debut Xenon Dreams. Favorite tracks include: Propofol (ft. Induction), Peripheral Access, Bradycardia
Reverse-phase high-pressure liquid chromatography
Well, I can think of two possible take homes from this data.
First the flippant one.
Don't prescribe Xanax.
Xanax isn't substantially different from other benzos.
Don't prescribe benzos.
Now the more serious one.
Anyone who thinks they can pin down the inevitably subjective effects of psychiatric drugs by citing the various objective biochemical performance indicators is pissing into the wind. The measure is how they effect the patient.
Most uncontroversially it's because pain and mood are the two symptom classes most susceptible to the placebo effect. Psychiatric drugs largely live or die on how effective they are as placebos. So you need to ask the person taking them, not the instruments measuring them in vivo or in vitro.
But perhaps more importantly, if only because it's a huge epistemological black hole at the centre of psychiatry into which few practitioners are prepared to gaze, the mind can neither be meaningfully reduced to chemical reactions nor to whatever subset of the attributes of those reactions we're able to quantify.
This isn't to say the mind isn't entirely emergent from physics and chemistry. I'm not flogging Cartesian dualism here. But the proximate and remote causes giving rise to it are so multitudinous and interact so complexly that imagining you can read them off the instruments of chemical analysis (or the symptom checklists in the DSM and ICD bibles) is not science. It's pseudoscience built on overextended, oversimplified scientism.
I've long been really puzzled that diazepam doesn't have a much longer duration of action. The fact that taking another dose the next seems to have very similar effects as the first dose makes me skeptical of a purely tolerance based effect.
I noticed the remark about diazepam disassociating from the receptors more quickly but I'm not sure I followed why/if that would explain why it's effects wear off so much more quickly than the half-life would predict. Is it somehow being stored in an unavailable form?